
Pharmacology Not Taught in Medic School: Succinylcholine Edition
- FOAMfrat
- Jan 31, 2022
- 6 min read

Why does Succinylcholine have a laundry list of contraindications? Why is it the neuromuscular blocker that induces fasciculations? What life-threatening condition can manifest as a result of Succinylcholine administration?
Keep reading for the next series of “Pharmacology not Taught in Medic School: Succinylcholine Edition”.
I. Mechanism of Action
a. The Neuromuscular Junction
Before we get to the specifics, we must set the playing field, the ground rules if you will. The site of action for our paralytics is the neuromuscular junction (NMJ), or the location where the neuron to stimulate muscle contraction interfaces with the actual muscle itself. The general anatomy of the NMJ looks like this:
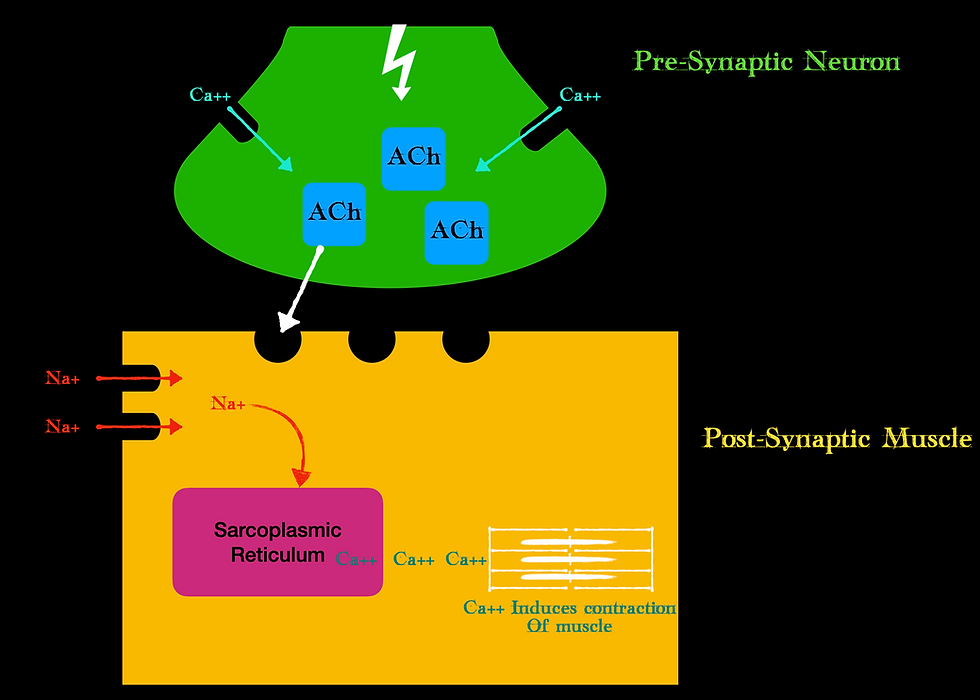
We have a presynaptic bulb where the neuron ends and a postsynaptic bulb where the muscle starts. Note, that the neuron does not technically touch the muscle, it is separated by that synaptic cleft. Our CNS sends a signal, or action potential down the neuron. This stimulates Calcium to be released at the pre-synaptic bulb. This calcium releases the neurotransmitter acetylcholine (ACh) from vesicles stored in the pre-synaptic bulb. The ACh is then released from the pre-synaptic neuron into the synaptic cleft where it binds to postsynaptic receptors. These postsynaptic receptors are sometimes called the motor endplate. Once ACh contacts the receptor, voltage-gated sodium channels open on the muscle and transmit this action potential to the inside of the muscle where further calcium is released from the sarcoplasmic reticulum. This calcium then participates in a process called excitation-contraction coupling where myosin proteins interact with actin proteins that produce the actual muscular contraction at the cellular level.
Not too bad right? Easy (enough) to follow! That is the playing field.
b. Succinylcholine hijacking
Succinylcholine is a molecule that looks very similar to acetylcholine. In fact, it is just two ACh molecules pieced together. So naturally, anywhere ACh goes, Succinylcholine (Sux for short) can go as well.

If ACh’s primary target is the receptors on the motor endplate, then once Sux hits our system, it acts on those same receptors. But since Sux is two ACh molecules glued together it actually has a higher affinity for those motor endplate receptors than ACh does. So Sux does not necessarily antagonize the receptor, it is a more potent agonist of the receptor and will beat out ACh every time (Vardanyan & Hruby, 2006).
The issue is that the motor endplate receptor does not distinguish ACh from Sux, the receptor just knows to either turn on or turn off based on the shape of the molecule that has attached to its receptor site. It is kind of like a cat, if it fits then it sits.
In this case, Sux attaches, the receptor just sees that a structurally fitting molecule has arrived and activates those voltage-gated sodium channels. Hence why we see some form of muscular action in the form of fasciculations when we administer Sux - It is all about chemical structure and function.
ACh is normally broken down and removed from the receptor by acetylcholinesterase. However, although Sux looks structurally like ACh, acetylcholinesterase does not degrade down Sux. So, it tends to linger on the receptor longer than usual. This desensitizes the receptor since nothing further can attach to it as Sux has taken up that spot – This is when paralysis takes place, and we can initiate our intubation (Open Anesthesia, n.d.).
So how does Sux eventually come off the receptor? It has its own enzyme called pseudocholinesterase that is produced by the liver (Trujillo & West, 2021). The issue is that this enzyme must mobilize itself and get to the area to do work, hence why Sux has such a short half-life. It eventually gets degraded but hopefully buys us enough time to facilitate an airway.
Remember, Sux looks like ACh, acts like ACh but does not get degraded like ACh. Instead, it lingers around a bit longer, causes some small muscular contractions (aka fasciculations), then eventually gets kicked off by its own degrading enzyme. That’s the short and skinny of it. All that work for 10 seconds of fun (sound familiar?)
II. Potassium Problems…
Most of the reason Sux has such a long list of contraindications is that Sux contributes to some degree of hyperkalemia. So, in essence, any condition that predisposes a patient to hyperkalemia, Sux is contraindicated (Smith MD, 2017).
As an average, typical RSI doses of Sux can increase serum potassium levels by 0.5 mEq/dL. Our hearts tend to be very sensitive to potassium so in theory, this “small” bump could be the difference between your run-of-the-mill peaked T wave and sine wave morphology (see Good Reads Episode 6). So how does Sux cause this increase in potassium?
Remember how at our NMJ, once ACh was attached to the motor endplate it activated voltage-gated sodium channels? This resulted in a net influx of Sodium and in exchange, the muscle cell shoved out potassium. This potassium that was once inside the cell now enters central circulation. There is simply an exchange of sodium for potassium at the post synapse.
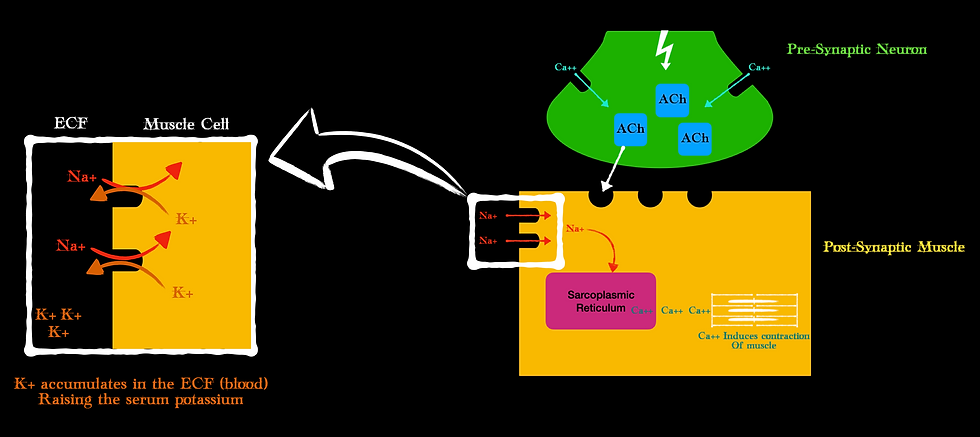
Here is something else to consider helping explain all these contraindications…
In both acute cases (i.e. trauma, crush injury, acidosis) and some chronic cases (i.e. neuromuscular disease, burns > 24 hrs), immature ACh receptors can proliferate in areas outside of the NMJ. This makes more receptors available for Sux to attach and do its damage to and ultimately facilitate more potassium into the central circulation.
Weird, I know…
Just remember, Sux contributes to hyperkalemia because of its action like ACh at the motor endplate. In acute and some chronic cases, these receptors can start popping up on the radar and be available for Sux to bind to and wreak havoc to our electrolyte balance.
III. A Magnificently Malignant Malady
If you work for an agency that allows RSI or you work in a setting where this procedure is done routinely then you are familiar with Malignant Hyperthermia (MH). MH is a condition of hypermetabolism and patients are genetically predisposed to it. There are a few known triggers and Sux is one of them. For providers, there may be the very rare chance you are the first to give a patient Sux so you will not know they have this genetic condition. Unless there is a familial history of issues related to anesthesia then I would probably assume this is the case and steer clear of Sux.
For those familiar with MH, interestingly, the clinical progression of MH tends to be quite similar. In fact, in 255 cases of MH, the clinical progression followed this timeline of events:
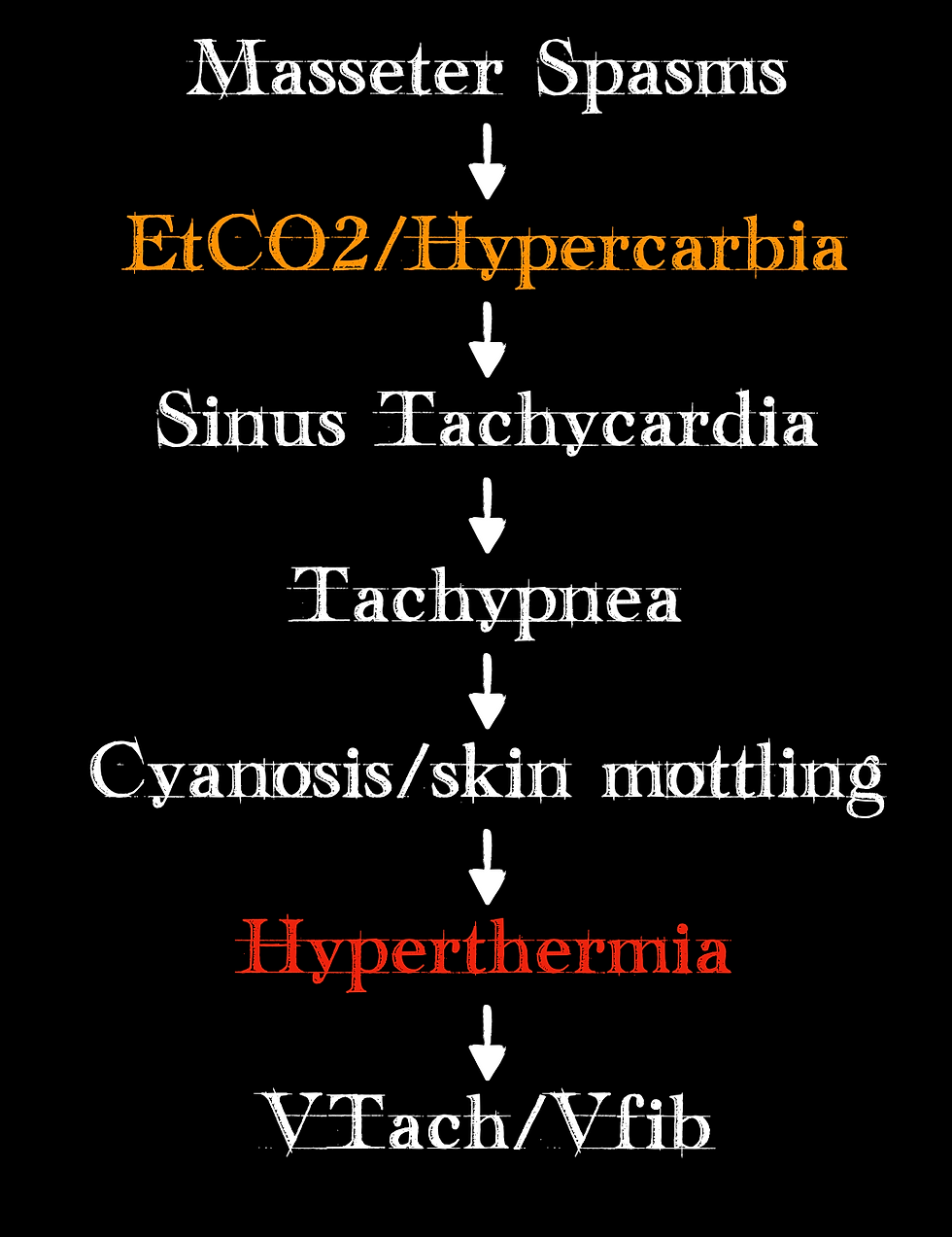
(York, 2019)
Note that a rise in end-tidal CO2 is the second item on the list! Something we should be monitoring and a change we should note, especially if we have not made any vent changes. Also, note that hyperthermia is closer to the bottom of the list than it is at the top of the list. If body temperature changes are the clinical finding we are relying on, we are already late in identifying that MH is occurring.
Now, my favorite part: the pathophysiology. MH is a hypermetabolic state characterized by sustained muscular contraction. Remember that after ACh binds to the motor endplate, the sodium channels open and eventually release calcium from the inside of the muscle at the sarcoplasmic reticulum. The calcium release is controlled by another receptor, known as the Ryanodine receptor. It sits on the surface of the sarcoplasmic reticulum and controls the flow of calcium.
MH is a genetic mutation of this Ryanodine receptor. Exposure to Sux causes this receptor to sit in its open state allowing a mass efflux of calcium out of the sarcoplasmic reticulum. Since calcium is required for muscle contraction (remember it facilitates the binding of myosin and actin – the contractile units of muscle), an excess of calcium means sustained contraction (Wappler, 2001).
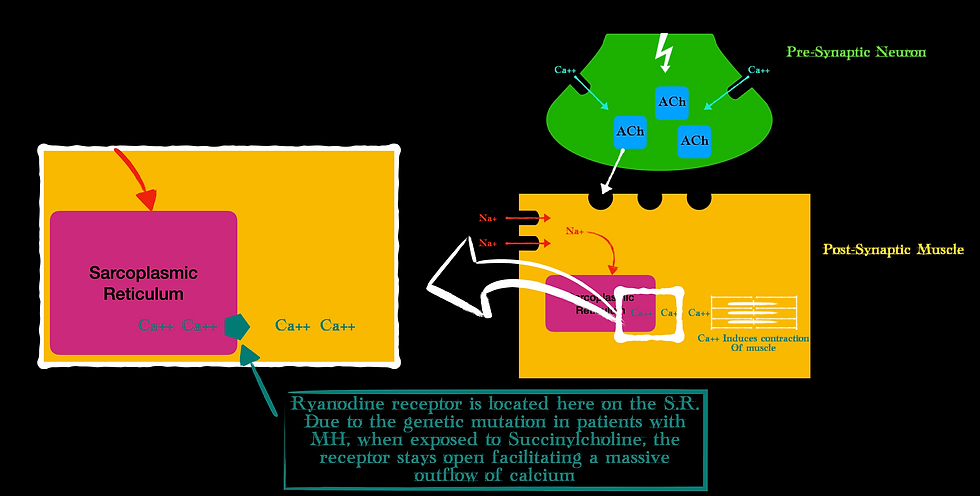
Now, ATP is required for the myosin and actin to detach (this is why rigor mortis happens, a dead person does not produce ATP so the myosin and actin don’t detach and we get stiff bodies). But the rate of calcium release because of the genetic mutation from MH on that Ryanodine receptor far exceeds the rate at which ATP can detach all those myosin and actin filaments.
Kind of like this guy making progress shuttling this water:
As our ATP gets burned up, we release CO2, we release heat, energy, etc. This all manifests in the signs and symptoms we see in the MH clinical progression.
Some final points to remember about our good friend Sux:
Sux is very structurally similar to ACh, so it behaves like ACh thereby producing fasciculations
Sux contributes to some degree of hyperkalemia because it still opens voltage-gated sodium channels in the motor endplate, and the muscle will “swap” potassium for incoming sodium.
Beware of Malignant Hyperthermia. It is genetic and you may be the first provider to encounter it. Watch that end-tidal CO2!

References
Open Anesthesia. (n.d.). Phase II depolarizing blockade. Www.openanesthesia.org. https://www.openanesthesia.org/aba_phase_ii_depolarizing_blockade/
Smith MD, C. (2017, October 16). Back to Basics: Succinylcholine and Hyperkalemia | EM Daily. Emdaily.cooperhealth.org. https://emdaily.cooperhealth.org/content/back-basics-succinylcholine-and-hyperkalemia
Trujillo, R., & West, W. P. (2021). Pseudocholinesterase Deficiency. PubMed; StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK541032/
Vardanyan, R. S., & Hruby, V. J. (2006). Synthesis of essential drugs. Elsevier, (Imp.
Wappler, F. (2001). Malignant hyperthermia. European Journal of Anaesthesiology, 18(10), 632–652. https://doi.org/10.1097/00003643-200110000-00002
York, B. C., PharmD, BCPS, BCCCP, BCIDP Critical Care Clinical Pharmacist, PGY-1 Pharmacy Residency Director UHS Hospitals Johnson City, New York Lyndsay Wormuth, PharmD, BCACP Ambulatory Care Clinical Pharmacist UHS Hospitals Johnson City, New. (2019, January 18). Malignant Hyperthermia: An Overview. Www.uspharmacist.com. https://www.uspharmacist.com/article/malignant-hyperthermia-an-overview